20.109(S21):M1D1

Contents
- 1 Introduction: Generate single-chain antibody fragment (scFv) library
- 2 Protocols:
- 2.1 Part 1: Investigate our parental scFv clone: plasmid Lyso_scFv_6
- 2.2 Part 2: Mutagenesis of scFv clone using Error-prone PCR
- 2.3 Part 3: Gel purify PCR products
- 2.4 Part 4: Prepare error-prone PCR products and backbone for yeast transformation
- 2.5 Restriction enzyme digest of pCTcon2 backbone
- 2.6 Part 5: Transform DNA into yeast
- 2.7 Part 6: Complete Orientation quiz
- 3 Navigation links
Introduction: Generate single-chain antibody fragment (scFv) library

To engineer a protein-protein interaction (antibody-antigen), we will use a method called directed evolution. Directed evolution is defined as an iterative process of mutating a gene, then screening those mutants for a particular functionality, collecting the best mutants and repeating. It is a method that is inspired by the process of natural selection. Directed evolution is very similar to Affinity Maturation, the process that results in higher affinity antibodies in the human immune system. This process is outlined in the figure to the right.
In the Wittrup lab protocol, successive rounds of random mutagenesis of the scFv gene, followed by expression of the scFv on the surface of yeast then selection for "good binders" by fluorescence activated cell sorting is carried out. This process will produce an antibody fragment with increased affinity or stability to its antigen.
Today we will walk through the technical details of random DNA mutagenesis via error-prone PCR. This process creates a pool of scFv genes that have random mutations throughout the DNA sequence. This collection of varied scFV genes are called a library.
Finally, we will outline how one integrates this library into an expression vector, also referred to as the backbone, vector or plasmid, that can then be transformed into yeast and expressed on the surface of the cells. This method is called cloning. We will also introduce integral tools when cloning DNA plasmids: polymerase chain reactions (PCR) and restriction endonucleases (RE).
Protocols:
Part 1: Investigate our parental scFv clone: plasmid Lyso_scFv_6
Motivation: To understand the important features of scFv yeast display it's important to know what genes are being utilized to control this system.

We will use SnapGene software for our DNA visualization and alignments. Feel free to use any software you are familiar with to carry out analysis. If you are off campus you must log into a VPN connection prior to opening the SnapGene application. Here is the link to the VPN download and installation instructions. You may need to update the MIT SnapGene license number if you have not opened the application recently. The new license information can be found here.
You can download Lyso_scFv_6 parental clone sequence here.
In your laboratory notebook, complete the following:
- Find and copy the forward and reverse primers in Part 2 on this page. Add them to this file and determine the size of the PCR product.
- Edit->Find->Find DNA sequence then Primer-> Add Primer.
- Highlight the features that start with Aga2p and end with the stop codon of the feature Myc and create a new feature named scFv.
- Create new feature: Features->Add Feature; Feature name: scFv; Feature Type: CDS (coding sequence)
- Click 'translate feature in sequence view' and click ok.
- At the bottom of the screen switch to feature view.
- Find the scFv feature. How many amino acids is the coding sequence product?
- List all the domains of this protein.
- Also while in plasmid features look up the following genes, list their function (under note) and describe their role in our experimental approach.
- What is TRP1?
- What is Gal1 promoter?
- What is AmpR?
Part 2: Mutagenesis of scFv clone using Error-prone PCR
Motivation: To introduce random DNA mutations into our scFv clone we will carry out error-prone polymerase chain reaction (PCR). We will use special bases in the PCR reaction that mimic DNA damage and can erroneously pair with incorrect bases and will result in changes in the scFv DNA sequence.

PCR amplification The applications of PCR (polymerase chain reaction) are widespread, from forensics to molecular biology to evolution, but the goal of any PCR is the same: to generate many copies of DNA from a single or a few specific sequence(s) (called the “target” or “template”).

In addition to the target, PCR requires only three components: primers to bind sequence flanking the target, dNTPs to polymerize, and a heat-stable polymerase to carry out the synthesis reaction over and over and over. DNA polymerases require short initating pieces of DNA (or RNA) called primers in order to copy DNA. In PCR amplification, forward and reverse primers that target the non-coding and coding strands of DNA, respectively, are separated by a distance equal to the length of the DNA to be copied. PCR is a three-step process (denature, anneal, extend) and these steps are repeated. After 30 cycles of PCR, there could be as many as a billion copies of the original target sequence.
Based on the numerous applications of PCR, it may seem that the technique has been around forever. In fact it is just under 30 years old. In 1984, Kary Mullis described this technique for amplifying DNA of known or unknown sequence, realizing immediately the significance of his insight. "Dear Thor!," I exclaimed. I had solved the most annoying problems in DNA chemistry in a single lightening bolt. Abundance and distinction. With two oligonucleotides, DNA polymerase, and the four nucleosidetriphosphates I could make as much of a DNA sequence as I wanted and I could make it on a fragment of a specific size that I could distinguish easily. Somehow, I thought, it had to be an illusion. Otherwise it would change DNA chemistry forever. Otherwise it would make me famous. It was too easy. Someone else would have done it and I would surely have heard of it. We would be doing it all the time. What was I failing to see? "Jennifer, wake up. I've thought of something incredible." --Kary Mullis from his Nobel lecture; December 8, 1993
Please watch this animation of PCR created by LabXchange: [PCR Animation]This will help answer questions for your lab notebook.
In your laboratory notebook, complete the following:
- Calculate the volume of the components that need to be added to this reaction. The final volume of all components in the reaction is 50uL.
- What cycle of the PCR reaction will we have amplified a PCR product of the correct size?
| Component, concentration | Final concentration | Volume |
|---|---|---|
| 10X Taq buffer | 1X | |
| 50mM MgCl2 | 2mM | |
| 2mM Forward primer | 10uM | |
| 2mM Reverse primer | 10uM | |
| 10mM dNTPs | 200uM | |
| 20uM 8-oxo-dGTP | 2uM | |
| 20uM dPTP | 2uM | |
| 100pg/uL template DNA | 13.3 pg/uL | |
| 5 units/uL Taq polymerase | 0.05 units/uL | |
| dH2O up to 50uL |
- Retrieve a 200uL thin-walled PCR tube and combine the components above.
- It is best practice to add water first and enzyme (Taq) last.
- The template DNA is our parental scFv clone, Lyso_scFv_6.
- Optimal primers have been determined experimentally by the Wittrup lab.
- Forward 5'-CGACGATTGAAGGTAGATACCCATACGACGTTCCAGACTACGCTCTGCAG-3'
- Reverse 5'-CAGATCTCGAGCTATTACAAGTCCTCTTCAGAAATAAGCTTTTGTTC-3'
- Amplify DNA on the thermocycler using the following program:
| Cycles | Denaturation | Annealing | Polymerization |
|---|---|---|---|
| 1 | 94°C for 3m | ||
| 2-11 | 94°C for 45s | 60°C for 30s | 72°C for 90s |
| 12 | 72°C for 10m |
Analysis of the error-prone PCR conditions described above (10 PCR cycles) has shown that one to nine amino acid mutations results per scFv gene. If desired, doubling the number of PCR cycles to 20 would result in three to fourteen mutations per scFv gene.
Part 3: Gel purify PCR products
Motivation: To separate the PCR products from the template DNA we will use gel electrophoresis. We can use gel purification to exploit the difference in size between the DNA template and PCR product.
Electrophoresis is a technique that separates large molecules by size using an applied electrical field and a sieving matrix. DNA, RNA and proteins are the molecules most often studied with this technique; agarose and acrylamide gels are the two most common sieves. The molecules to be separated enter the matrix through a well at one end and are pulled through the matrix when a current is applied across it. The larger molecules get entwined in the matrix and are stalled; the smaller molecules wind through the matrix more easily and travel farther away from the well. The distance a DNA fragment travels is inversely proportional to the log of its length. Over time fragments of similar length accumulate into “bands” in the gel. Higher concentrations of agarose can be used to resolve smaller DNA fragments.

DNA and RNA are negatively charged molecules due to their phosphate backbone, and they naturally travel toward the positive electrode at the far end of the gel. Today you will separate DNA fragments using an agarose matrix. Agarose is a polymer that comes from seaweed. To prepare these gels, agarose and 1X TAE buffer (Tris base, acetic acid, and EDTA) are microwaved until the agarose is melted and fully dissolved. The molten agar is then poured into a horizontal casting tray, and a comb is added. Once the agar has solidified, the comb is removed, leaving wells into which the DNA samples can be loaded.
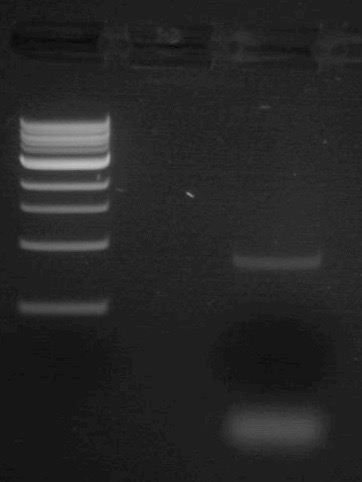
To purify the PCR mutant product from the parental template, a 1% agarose gel with SYBR Safe DNA stain was used to separate the DNA fragments. In addition, a well was loaded with a molecular weight marker (also called a DNA ladder) to determine the size of the fragments.
To ensure the steps included below are clear, please watch the video tutorial linked here: [DNA gel electrophoresis]. The steps are detailed below so you can follow along!
- Add 8 μL of 6x loading dye to 48uL of PCR product mix.
- Loading dye contains bromophenol blue as a tracking dye, which enables you to follow the progress of the electrophoresis.
- Glycerol is also included to weight the samples such that the liquid sinks into well.
- Flick the eppendorf tubes to mix the contents, then quick spin them in the microfuge to bring the contents of the tubes to the bottom.
- In the far left lane load 10 μL of 1kb DNA ladder then load 50 μL of the PCR/loading dye mix in a lane to the right.
- Run the samples at 125V for 45min.
- Image gel on the UV gel doc station.
- After taking an image of the PCR product on the UV gel doc station, the gel is moved to a bench top UV light source.
- Using appropriate safety equipment, the gel is illuminated by the bench top UV light source and the visible PCR product is cut out of the gel with a razor and stored in a micro centrifuge tube.
- The PCR product DNA is purified from the gel using the Qiagen QIAquick Gel Extraction Kit.
In your laboratory notebook, complete the following:
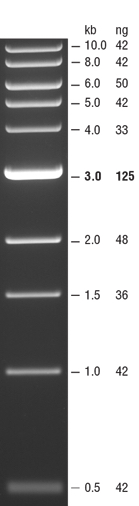
- Upload the following image of the gel to your lab notebook. Note the NEB 1kb ladder sizes here and estimate the size of the PCR product based off the gel image.
- Based off the sequence analysis you carried out in Part 1, is this result what you expected? Why or why not?
- Why is it important that we gel purify this PCR product? What two components of the first PCR reaction do we need to purify our product away from? Hint: They are not present in the second PCR reaction.
{kind=link}
{kind=link}
Part 4: Prepare error-prone PCR products and backbone for yeast transformation
Motivation: To transform the mutated scFv products and the plasmid backbone in yeast we must carry out two additional steps. First we will amplify the PCR products so there is a sufficient amount of DNA for transformation and second we will digest the backbone plasmid so the new mutated scFv genes can integrate into the plasmid.

- Combine the following components in a 200uL PCR tube.
- Multiple tubes can be prepared to yield more DNA for the following steps.
| Component, concentration | Final concentration | Volume |
|---|---|---|
| 10X Taq buffer | 1X | 10uL |
| 50mM MgCl2 | 2mM | 4uL |
| 10uM Forward primer | 2mM | 5uL |
| 10uM Reverse primer | 2mM | 5uL |
| 10mM dNTPs | 200uM | 2.0uL |
| Gel purified PCR product | 4uL | |
| 5 units/uL Taq polymerase | 0.05 units/uL | 1uL |
| dH2O | 69uL |
- Amplify the DNA using the following thermal cycler conditions:
| Cycles | Denaturation | Annealing | Polymerization |
|---|---|---|---|
| 1 | 94°C for 3m | ||
| 2-31 | 94°C for 45s | 60°C for 30s | 72°C for 90s |
| 32 | 72°C for 10m |
In your laboratory notebook, complete the following:
- What components were used in the PCR reaction in Part 2 that are not used here?
- What is the major difference in the thermocycler conditions? What do you think is the reason for this?
- Consider the following sequence: 5'-GG*G*CTT-3', where G* represents 8-oxo-G.
- If 8-oxo-G can pair with C or A, how many different dsDNA pairings are possible for this sequence? What are the base pair combinations?
- After one round of PCR what are all of the possible replication products from the sequence above?
Restriction enzyme digest of pCTcon2 backbone

Restriction endonucleases, also called restriction enzymes, 'cut' or 'digest' DNA at specific sequences of bases. The restriction enzymes are named according to the prokaryotic organism from which they were isolated. For example, the restriction endonuclease EcoRI (pronounced “echo-are-one”) was originally isolated from E. coli giving it the “Eco” part of the name. “RI” indicates the particular version on the E. coli strain (RY13) and the fact that it was the first restriction enzyme isolated from this strain.
The sequence of DNA that is bound and cleaved by an endonuclease is called the recognition sequence or restriction site. These sequences are usually four or six base pairs long and palindromic, that is, they read the same 5’ to 3’ on the top and bottom strand of DNA. For example, the recognition sequence for EcoRI is 5’ GAATTC 3’ (see figure at right). EcoRI cleaves the phosphate backbone of DNA between the G and A of the recognition sequence, which generates overhangs or 'sticky ends' of double-stranded DNA.
You can download the pCTCON2 backbone, or vector, plasmid here
To cut this circular plasmid and allow the incorporation of your mutant scFv library PCR product, the following restriction sites will be used: NheI and BamHI. Each restriction enzyme has particular buffer, temperature, and time condition for optimal DNA digest. When using more than one enzyme you must determine the best conditions for both enzymes. To determine the conditions of this digest enter both restriction enzymes into the NEB Restriction enzyme tool
In your laboratory notebook, complete the following:
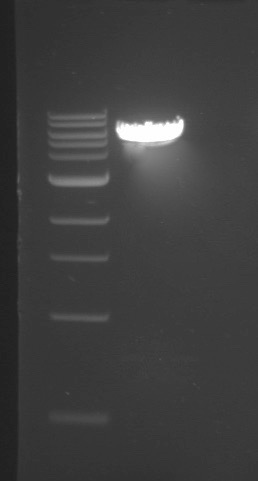
- What is the buffer, time and temperature for this reaction if you need to digest 1ug of pCTcon2 plasmid DNA?
- Upload the following image of the gel to your lab notebook. Note the NEB 1kb ladder sizes here and estimate the size of the plasmid DNA digest product based off the gel image.
- Open the pCTcon2 sequence in Snapgene and determine the size of the backbone after restriction enzyme digest. Is the gel image result expected? Why or why not?
{kind=link}
Part 5: Transform DNA into yeast
Motivation: To express the new scFv library we must transform the PCR product and digested backbone DNA into the yeast cells and select for re-circularized plasmid DNA.
We will use a transformation procedure called electroporation to get the DNA inside yeast cells. During this procedure yeast, specialized salt buffers, and DNA are loaded into a metal cuvette and a high voltage electrical pulse is used to open pores within the cell membrane. DNA is taken into the yeast cells and the pores close within minutes as the cells recover in full nutrient media.
- Inoculate 5mL of YPD yeast media with an EBY100 colony from a YPD plate and grow overnight at 30°C, shaking.
- The next day inoculate 50mL culture of YPD with the overnight liquid culture of EBY100 and grow at 30°C, shaking till yeast are in log phase growth (~6hrs.)
- Add 500uL Tris-DTT buffer to the liquid culture and continue to grow at 30°C, shaking for 15min.
- Pellet the yeast at 2,500g for 3min at 4°C, aspirate the supernatant and wash with 25mL ice-cold E buffer (resuspend in buffer, pellet, and aspirate supernatant) then repeat with 1mL ice-cold E buffer and finally 300uL of ice-cold E buffer.
- Prepare the DNA from Part 4 during these washes: Open 2 sterile electroporation cuvettes, to the first add 5ug of PCR product DNA (scFv gene library) and 1ug of RE digested pCTCON2 backbone. To the second add 1ug of RE digested pCTCON2 backbone alone, this is the negative control.
- Add 50uL of yeast resuspended in buffer E to the electroporation cuvettes and mix with DNA.
- Load one electroporation cuvette into gene pulser and electroporate at 0.54kV.
- Immediately add 1mL of warm (30°C) YPD to the cuvette and transfer cells to a 15mL tube. Wash cuvette with an additional 1mL of YPD to retrieve all cells.
- Shake cells at 30°C for 1 hour to allow for recovery.
- Pellet cells at 2,500g for 5min and remove supernatant.
- Resuspend cells in 100-1000mL of SDCAA media and allow growth at 30°C, shaking for 24-48rs.
- selection for yeast with TRP1 gene in stable, circular plasmid
Part 6: Complete Orientation quiz
Complete the orientation quiz with your partner. Though you are working with your partner, each student should record all answers independently. If you disagree with your partner on an answer, you should write what you think is the correct answer on your quiz.
Good luck!
Next day: Enrich candidate clones from library using FACS