20.109(S21):M1D2

Contents
Introduction: Enrich candidate clones from library using FACS (FACS)
Flow cytometry is a cell analysis technique that has revolutionized biology by allowing researchers to analyze and isolate cells based on their physical and fluorescent qualities. Genetic reporters, labeled antibodies and small molecule tags or dyes may be used to introduce fluorescent signal into cells. You can isolate a pure sample of a particular cell type from a complex mixture of cells by using a modified flow cytometer equipped to perform FACS: Fluorescence Activated Cell Sorting. If the machine is only able to count, and not sort subpopulations of cells, then the procedure is called flow cytometry, while cell sorting is called FACS.
Today we will be using FACS to sort a library of yeast, a library is a collection of DNA fragments, usually on plasmids, that is stored and propagated in a population of microorganisms. A single microorganism carting one plasmid in the library is called a clone. These yeast are expressing a single-chain antibody fragment (scFv) that binds to the enzyme lysozyme. Lysozyme is an abundant, inexpensive and commercially valuable enzyme for the food and pharmaceutical industry. Our goal is to find a clone of a single-chain antibody fragment that has improved binding to lysozyme over the parental clone Lyso_scFv_6. We will use fluorescently labeled antibodies to mark yeast that express the scFv and fluorescently labeled streptavidin to mark the presence of bound lysozyme. Using these two fluorescent markers together we will isolate a population of yeast from the library that bind to lysozyme with high affinity.

Fluorescence Activated Cell Sorters and flow cytometers have been around since the 1970s, and while the details are fancier (more colors, faster measurements, etc), the general principles have remained the same. It is truly a testament to the versatility and utility of flow cytometry that the technology does not look to be supplanted by another approach any time soon.
Protocols:
Motivation: To screen the new scFv library for improved binders to the antigen, lysozyme, we must incubate the yeast with lysozyme then use fluorescent antibodies to quantify the scFv expression and lysozyme bound.
The library of yeast you will be sorting today were started from a population of cells that was tenfold the estimated starting library diversity (estimated number of unique clones generated) to assure we will have adequate coverage of the single-chain antibody fragment clones represented in this population. The single-chain antibody fragment (scFv) expression was induced by growing the EBY100 yeast transformed with the plasmid library in SGCAA media (containing galactose) overnight at 20°C. The galactose in the media activated expression of proteins under the GAL 1 promoter and the lower temperature 20°C rather than 30°C assures proper protein folding.
Part 1: Prepare cells for cell sorting

- Obtain an aliquot of induced yeast library from the 4°C cooler and vortex to resuspend the cells.
- Measure the optical density of the yeast at 600nm using the spectrophotometer.
- Remember to bring media alone as a blank.
- An optical density of 1 at 600nm equals 1x107yeast per mL.
- Calculate the volume needed to have 1x107yeast and add that volume of yeast suspension to a 1.5mL tube. In the tube with 1x107 yeast bring the volume to 1mL of PBSA.
- For this experiment you will need a total of five 1.5mL microcentrifuge tubes with 1x107yeast in each tube.
- Cell sorting experimental conditions and controls:
- (1) Library of yeast with 5nM lysozyme/complete labeling
- (2) Parental clone Lyso_scFv_6 with 5nM lysozyme/complete labeling
- (3) Library with no label control
- (4) Library with AF488 only control
- (5) Library with AF647 only control
- Cell sorting experimental conditions and controls:
- Pellet the yeast at 7,500g for 30sec and remove the supernatant.
- Resuspend the yeast pellet in 1mL fresh PBSA. Pellet the yeast once more and remove the supernatant.
- Resuspend the yeast in Condition 1 and 2 with 1mL fresh PBSA with 5nM of biotinylated lysozyme. All other controls (conditions 3-5) are resuspended with 1mL PBSA.
- The stock of biotinylated lysozyme is 1.87mg/mL and the molecular weight is 14,300. Calculate the volume of stock to dilute into 1mL of PBSA to make a final concentration of 5nM.
- Incubate condition 1 and 2 (yeast/lysozyme mix) for 120min at room temperature on the nutator. Same conditions for controls 3-5.
- After 60min add 1:1000 anti-c-myc primary antibody to condition 1 and 2 (yeast/lysozyme mix), invert 3 times and place the mix back on nutator for an additional 60min at room temp.
- Pellet conditions 1-5 at 7,500g for 30sec and wash the cells with 1mL ice-cold PBSA.
- Resuspend the cells in condition 1 and 2 in 500uL of ice-cold PBSA with 1:1000 AF488 goat anti-chicken IgG secondary antibody and 1:1000 AF647 conjugated to Streptavidin.
- Resuspend the cells in condition 3 in 500uL of ice-cold PBSA alone, condition 4 resuspended in 500uL of ice-cold PBSA with 1:1000 AF488 goat anti-chicken IgG secondary antibody alone, and condition 5 in 1:1000 AF647 conjugated to Streptavidin alone.
- Leave all tubes protected from light and on ice for 30min.
- Pellet the cells and resuspend in 1mL of ice-cold PBSA. Leave on ice until cell sorting.
In your laboratory notebook, complete the following:
- After transformation of the library into yeast we carried out a titration on agar plates to determine how many clones were taken up into yeast. From this experiment we estimate 1x106 clones are in our library. How many cells did we use to start our culture to ensure full coverage of our library diversity?
- If the optical density of the overnight yeast culture is OD@600nM=2.2, what volume of cells equals 1x107yeast?
- The stock of biotinylated lysozyme is 1.87mg/mL and the molecular weight is 14,300. Calculate the volume of stock to dilute into 1mL of PBSA to make a final concentration of 5nM.
Part 2: Fluorescence Activated Cell Sorting (FACS)
Motivation: To harvest the yeast cells expressing scFvs with improved binding to lysozyme we must quantify the binding capacity and separate out improved binders via Fluorescence Activated Cell Sorting.
Cell sorting, or FACS, is technically challenging and most FACS machines are only run by experts. This analysis will be done by Sarah, a trained PhD student in the Wittrup lab, and we will describe the main steps to cell sorting during the video and they are detailed below.
To ensure the steps included below are clear, please watch the video tutorial linked here: FACS of yeast candidate clones bound to lysozyme.
- Place autoclaved or sterile tubes into the FACS collection rack with 1mL of PBSA in each tube.
- Set-up sorting gates using control yeast prepared alongside the library staining.
- Next, quickly vortex the stained yeast library and forcefully pipette the cell through the 35µm nylon mesh cap of a Falcon flow cytometry tube.
- This reduces the likelihood of cell aggregates, which can cause critical errors and clogs in the machine.
- Immediately place the cells on the sample injection port (SIP.)
- Cells will begin to flow through the machine. They are separated into a single cell stream, pass through analyzer and are charged, then sorted into the collection tubes. In our experiment we are only collecting one population the scFv yeast with increased binding affinity to lysozyme.
- For this experiment, once the controls have established a gate, or population of desired cells, the sort of the 1x107yeast takes about 20minutes.
- Once the sorting is complete, rinse the tube with 1ml SDCAA and put on ice.
- Transfer the cells back to lab and add additional SDCAA media to bring the volume up to 5mL.
- Leave the cells at 30°C for 2-3 days.


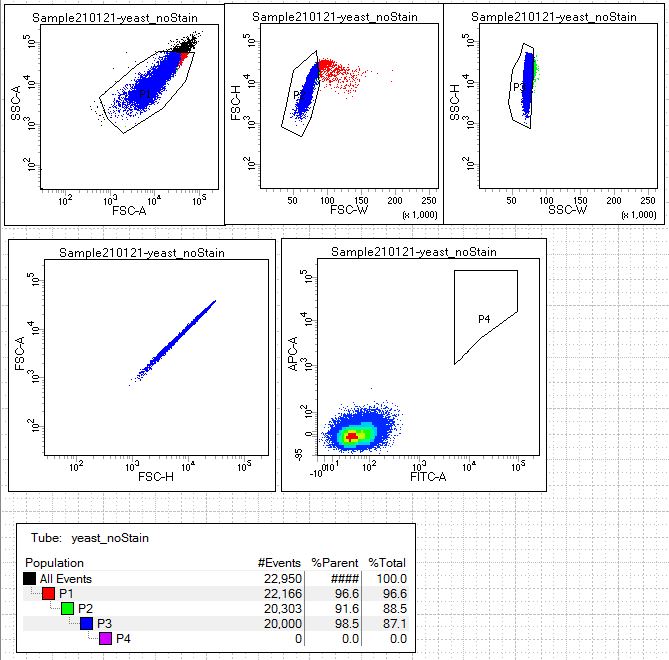
The key to collecting the population of cells you would like is running the appropriate controls to identify these cells accurately on the FACS machine. The first control to analyze is the mock/negative control; for our experiment this is unstained yeast (condition 3). Open the scatterplots for this control in the following link: no label control.
{kind=link}
Note that this data is represented in a density scatterplot, meaning the highest density of cells is red and lowest density of cells is blue. The red/yellow/green/blue hot spots indicate increasing numbers of events resulting from discrete populations of cells. The density plot representation enables grouping of the peaks and valleys that distinguish subsets of cells.
These cells are first analyzed using side scatter versus forward scatter after the cell passes through the analysis point of the laser. Forward scatter (FSC) is a measurement of the sample's refraction of light and therefore often is correlated with cell size. Side scatter (SSC) tells us something about internal complexity of the cell, like size of nucleus or other organelles. The cells should be the largest objects in the solution, while dust and cell debris will fall at the lower left of the scatter axis. Cell aggregates are common and will appear in the top right of the scatter axis. Experienced users can delineated gates to separate single yeast cells from doublets or aggregates. Gates are drawn or selected by the user in the software and select cell populations with particular characteristics, like size or fluorescent labels. Gates P1, P2, and P3 are drawn to select for single yeast cells and exclude debris or cell aggregates.
In last plot on the lower right, the cells are analyzed by the fluorescent signal from the laser measurements. This BDFACS Aria II is configured as a two laser system, a Blue laser (488nm) and a Red laser(633nm). We must choose the optical filters that are appropriate for our fluorophores attached to our secondary antibody and antigen label. For this system we will use the FITC-A and APC-A filters. The FITC filter measures AF488, detecting our scFv expression, and the APC filter detects AF647, detecting the presence of the ligand. We'll talk more about filter selection later in the module but if you'd like to consider the fluorophore and filter options the BD Spectral Viewer BD Spectrum Viewer is a great tool. The unstained yeast have no fluorophores associated with the cells so we should see a single population of cells in the lower left of the FITC v. APC plot that represents no fluorescent signal.
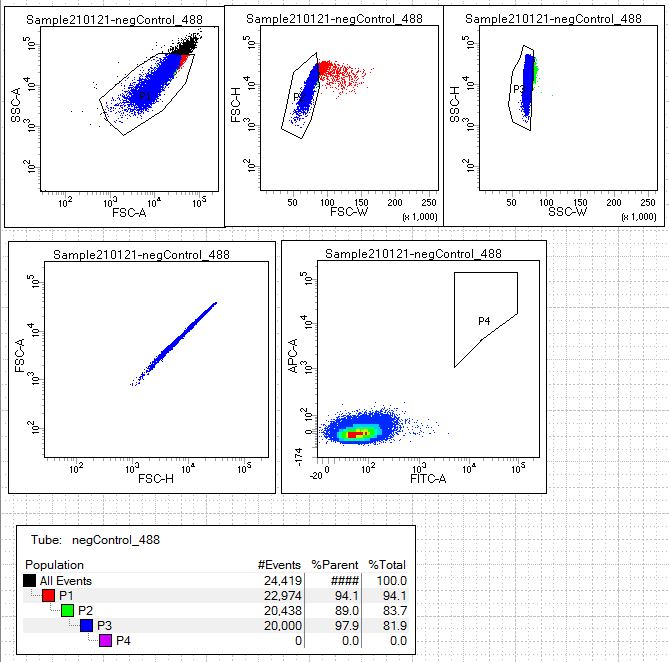
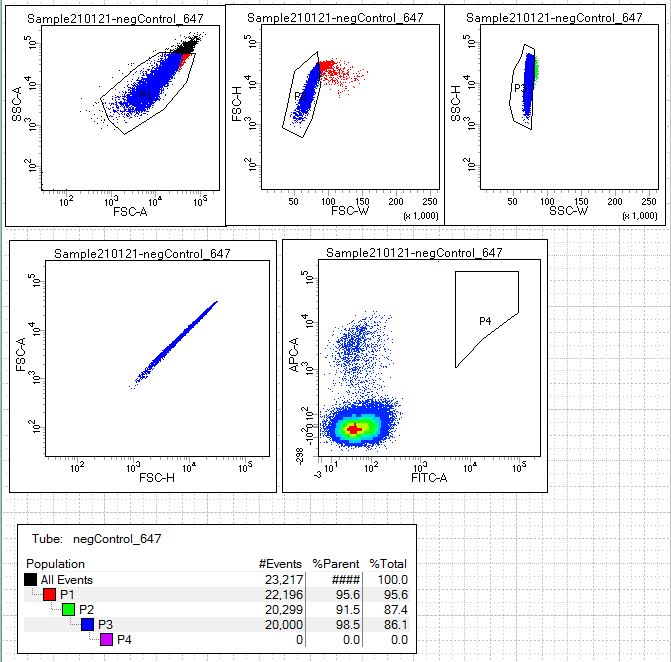
Next we look at our single-color fluorescence negative staining controls (conditions 4 and 5). Open the scatterplots for these controls in the following links: 488neg control and 647neg control.This allows the user to establish the signal from that single fluorophore that might be nonspecifically sticking to the yeast expressing scFv. These samples will be yeast stained with secondary antibodies conjugated to fluorophore AF488(green) alone and yeasted stained with streptavidin conjugated to AF647(far-red) alone. If a population of the yeast library are binding nonspecifically to the secondary antibody or streptavidin these cell populations should be excluded from the sort.
{kind=link}
{kind=link}
Finally, we look at our positive control (condition 2), the previously characterized clone Lyso_scFv_6. Open the scatterplot for this control in the following link: positive control. This allows the user to establish the population of cells that express scFv and bind to lysozyme. These samples will be yeast stained with antibodies conjugated to fluorophore AF488 (green) and with streptavidin conjugated to AF647 (far-red).
{kind=link}
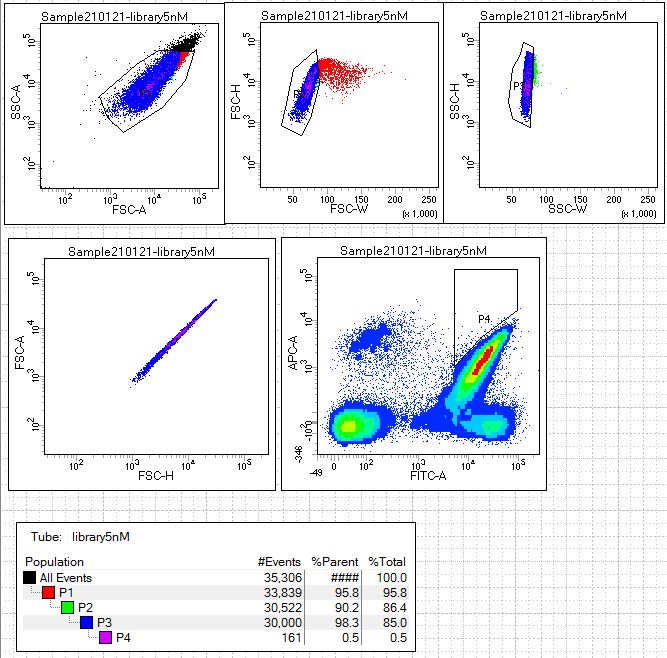
By collecting the cells that have higher green and higher far red signal than our parental clone we hope to identify scFv clones that have improved lysozyme binding during the analysis and sorting of the newly generated scFv library (condition 1). Open the scatterplot for the experimental sort in the following link: library sort with 5nM lysozyme.
{kind=link}
In your laboratory notebook, complete the following:
- What is a density scatterplot? What do the colors represent?
- What is an event?
- What is a gate?
- The plots for no label control or condition 3 is where gates P1, P2 and P3 are set. What events are we trying to exclude using these gates? What events are we trying to exclusively include?
- Look at the scatterplots for negative control 488 and 647 (condition 4 and 5). Did we expect these to look different from control 3? Which fluorophore has non-specific signal (488 or 647)? Explain your observation.
- The parental or positive control (condition 2) is used set and adjust gate P4. What do you notice about the FSC-H v. FSC-W plot? What does this suggest?
- The scatterplot analyzing the library sort (condition 1) represents diverse set of yeast (not just one clone like the positive control plots). Describe the four major groups: lower left quadrant, upper left quadrant, lower right quadrant and finally the yeast in gate P4, upper right quadrant.
- In this experiment we are measuring scFv binding to lysozyme at an equilibrium state. Why did we choose to incubate with 5nM lysozyme for the cell sorting?
- We also carried out this experiment by incubating 100nM lysozyme but did not share this data? Would we expect to see more or fewer yeast in our P4 gate population when cells are incubated with 100nM lysozyme?
Part 3: Participate in group paper discussion
During lab we will review and discuss Boder, E. T., et al. "Yeast surface display for screening combinatorial polypeptide libraries" Nature Biotechnology. (1997) 15:553-557.
From the Introduction
Consider the key components of an introduction:
- What is the big picture topic?
- Is the importance of this research problem clear?
- Are you provided with the information you need to understand the research?
- Do the authors include a preview of the key results?
From the Results and Discussion
Carefully examine the figures. First, read the captions and use the information to 'interpret' the data presented within the image. Second, read the text within the results section that describes the figure.
- Do you agree with the conclusion(s) reached by the authors for Figure 2?
- What controls were included and are they appropriate for the experiment performed represented by Figure 2?
- Are you convinced that the data are accurate and/or representative for Figure 2?
In this paper the results are interpreted and discussed as they are described in the text:
- Are the results summarized after they are described?
- Do the authors overreach when interpreting the data?
- Did the authors 'tie' the data together into a cohesive and well-interpreted story? This is usually the last paragraph of a research article.
- Are the data linked back to the big picture topic from the introduction?
In your laboratory notebook, complete the following:
- Based on your reading and the group discussion of the article, answer the bulleted questions above.
- What is one take away message from Figure 3?
- What is the difference between panel A and panel B in Figure 4?
- What the measured difference between mutants 4M1.1 and 4M1.2 and the wild-type (or parental) scFv in Figure 5? What does this mean in terms of scFv-antigen binding?
Reagents
- SGCAA media: 2% Galactose, 0.67% Yeast Nitrogen Base, 0.5% Casamino Acids, 0.54% Na2HPO4, 0.86% NaH2PO4 then sterilized (Sigma)
- SDCAA media: 2% Dextrose, 0.67% Yeast Nitrogen Base, 0.5% Casamino Acids, 0.54% Na2HPO4, 0.86% NaH2PO4 then sterilized (Sigma)
- PBSA: Phosphate buffered saline, 0.1% albumin (Sigma)
- Biotin lysozyme from chicken egg white (Sigma)
- anti-c-myc, chicken IgY fraction (ThermoFisher)
- Alexa Fluor 488 goat anti-chicken IgG (ThermoFisher)
- Alexa Fluor 647 conjugate streptavidin (ThermoFisher)
- BD FACS Aria II instrument
- Falcon flow cytometry filter cap tube (Fisher Scientific)
Next day: Harvest clone plasmids from library
Previous day: Generate scFv library